
Нейрофіброматоз
(Neurofibromatosis)
Наталія Афанасьєва
Завідуюча Кримським республіканським медико-генетичним центром,
кандидат медичних наук
Нейрофіброматоз відноситься до групи факоматозів (грецькою phakos – пляма, matosis – пухлина). Сьогодні ця назва поєднує кілька захворювань, які насправді є різними нозологічними формами: нейрофіброматоз I, II, III і IV типів. Біля 90% усіх хворих на нейрофіброматоз мають нейрофіброматоз I типу.
Нейрофіброматоз I типу (НФ1, класичний, периферійний, власне хвороба Реклінгхаузена, номер в OMIM – 162200) — це системне спадкове захворювання з переважним ураженням шкіри та нервової системи, успадковується аутосомно-домінантно, з високою пенетрантністю і варіабельною експресивністю. Захворювання обумовлене мутацією гену “NF1” в 17q-хромосомі. Чоловіки та жінки уражуються однаково часто.
Нейрофіброматоз був описаний німецьким патологом Фрідріхом Даніелем фон Реклінгхаузеном (Recklinghausen F.D.) в 1882 році в роботі “Про множинні фіброми шкіри та їх зв’язок з множинними невромами”, після цього захворювання отримало його ім’я.
НФ1 – це одне з найпоширеніших моногенних захворювань людини, зустрічається з частотою не рідше 1:3000 — 1:4000 населення. Загальна кількість хворих у світі наближається до 1 мільйону. В Україні кількість хворих становить приблизно 10-11 тисяч (достовірні дані про частоту цього захворювання в нашій країні відсутні).
Етіологія та патогенез:
Ген NF1 локалізується на довгому плечі 17 хромосоми (17q11.2). Приблизно половина випадків — це наслідок нових мутацій. Частота мутацій гену NF1 визначена як 1 х 1014 (одна з найвищих серед спадкових захворювань людини). При цьому захворюванні описані різні типи мутацій: великі та малі делеції, транслокації гену, заміни нуклеотидів (всього більше 100 варіантів), що значно ускладнює їх пошук під час молекулярно-генетичного дослідження.
НФ1 має повну (100%) пенетрантність, тобто усі носії патологічного гену є хворі, але експресія гену, тобто ступінь викликаних ним клінічних проявів, дуже варіюється. Навіть в межах однієї сім’ї можуть спостерігатися як мінімальні прояви, так і важкі випадки.
Ген NF1 є одним з основних генів-супресорів пухлинного росту для 30% тканин організму, в першу чергу нейроектодермального походження. Продуктом гену NF1 є великий білок нейрофібромін, який забезпечує контроль за ростом клітин. При пошкодженні цього гену в одній з хромосом 17-ої пари 50% нейрофіброміну, що синтезується, стає дефектним і спостерігається зміщення рівноваги росту клітин в бік проліферації та/або недостатньої диференціації клітин, що зрештою призводить до розвитку пухлин.
Захворювання має виражений клінічний поліморфізм, прогресуючий перебіг, поліорганність уражень та високу частоту ускладнень (злоякісне переродження нейрофібром, судомні напади тощо).
Діагностичні критерії НФ1:
Клінічна діагностика НФ1 ґрунтується на виявленні діагностичних критеріїв, рекомендованих Міжнародним комітетом експертів з нейрофіброматозу при Національному інституті здоров’я США в 1987 році. Діагноз може бути поставлений при наявності у хворого не менше двох з перелічених нижче ознак:
- Не менше п’яти плям кольору “кави з молоком” діаметром більше 5 мм у дітей препубертатного віку і не менше шести таких плям діаметром більше 15 мм в постпубертатному віці.
- Дві та більше нейрофіброми будь-якого типу чи одна плексиформна нейрофіброма.
- Множинні дрібні пігментні плями по типу веснянок, які локалізуються у великих складках шкіри (під пахвами та/або в паху) – симптом Кроува (Crowe).
- Гліома зорового нерву.
- Два та більше вузликів Ліша на райдужній оболонці, їх можна побачити при огляді за допомогою щілинної лампи.
- Дисплазія крила клиноподібної кістки або вроджене потоншання кортикального шару довгих трубчастих кісток з наявністю псевдоартрозу чи без нього.
- Наявність у родичів першого ступеня нейрофіброматозу I типу за тими ж критеріями.
Особливістю захворювання є специфічна послідовність проявів симптомів в залежності від віку пацієнта, що ускладнює клінічну діагностику НФ1 в ранньому дитячому віці. Таким чином, від народження та протягом перших років життя можуть існувати лише деякі ознаки нейрофіброматозу I типу, наприклад, великі пігментні плями, плексиформні нейрофіброми, скелетні дисплазії. Інші симптоми можуть проявлятися значно пізніше (в 5–15 років). При цьому виразність клінічних проявів, протікання та швидкість прогресування НФ1 у різних хворих неоднакові і коливаються в широких межах.
Найчастіше трапляються прояви з боку шкіри. Пігментні плями чітко окреслені, світло-коричневого кольору, можуть зустрічатися при народженні, проте найчастіше з’являються протягом першого року життя (у 82% випадків). До 4-го року життя (або раніше) вони реєструються в усіх дітей з НФ1 (фото 1).
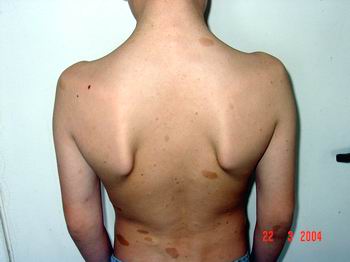
Нейрофіброми (дермальні, гіподермальні, плексиформні) є найвиразнішим проявом хвороби Реклінгхаузена, їх кількість іноді сягає кількох сотень (фото 2).
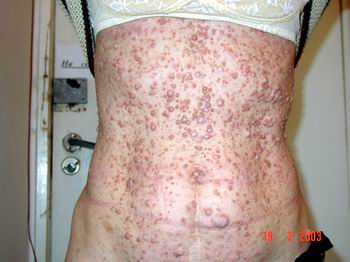
Нейрофіброми при народженні зустрічаються рідко, частіше вони з’являються у 3-5 років. Час від часу їх кількість і розміри зростають внаслідок дії різних стимулюючих факторів, серед яких найважливішими є гормональна перебудова організму: пубертатний вік, період вагітності та після пологів, а також перенесені травми або тяжкі соматичні захворювання. Із збільшенням спектру пропонованих комерційних медичних та косметичних послуг значно збільшилась кількість звернень хворих, які вказують на появу нових пухлин (нейрофібром, неврином, шванном) після ятрогенних втручань. Мова йде про видалення пухлин з діагностичною або лікувальною метою за допомогою різних методів, включаючи хірургічне вирізання. До ятрогенних ускладнень також призводить призначення фізіотерапевтичних процедур при лікуванні різних соматичних захворювань, корекція скелетних порушень (різноманітних видів сколіозу, переломів) та нервово-м’язових розладів (дуже часто дітям грудного віку призначається масаж, коли діагностика нейрофіброматозу I типа неможлива через недостатність клінічних проявів). Але часто захворювання прогресує і на фоні гаданого благополуччя.
Плексиформна нейрофіброма являє собою м’яку рихлу масу, яка „повзе” по ходу нерва, як правило, трійчастого або верхнього цервікального, іноді з інтенсивним коричневим забарвленням, і яка є значним косметичним дефектом (фото 3). Плексиформні нейрофіброми можуть бути гігантськими, масою більше 10 кг, часто вони пов’язуються з підвищеним ризиком переродження у злоякісні пухлини. При локалізації в середостінні, в черевній порожнині, в очній ямці вони призводять до порушення функцій прилеглих органів. Зустрічаються приблизно у 4% хворих.
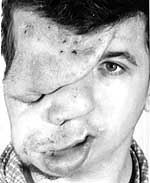
Гістологічно нейрофіброми складаються з фібробластів, клітин Шванна, периневральних клітин. Крім того, в складі нейрофібром є велика кількість тучних клітин. При електронній мікроскопії спостерігається їх тісний контакт з лімфоцитами.
Симптом Кроува (Crowe) — дифузна пігментація (по типу веснянок) під пахвами або в крупних складках (фото 4). Симптом зустрічається у 70% хворих на НФ1, як правило, починаючи з середнього дитячого віку.
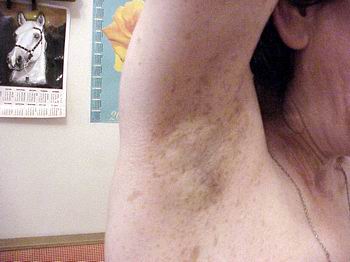
Вузлики Ліша – це пігментована меланоцитна гамартома райдужної оболонки, яку знаходять при огляді за допомогою щілинної лампи. У випадку множинних вузликів цей симптом є патогномонічний для НФ1 і спостерігається у більш ніж 90% хворих протягом другого десятиріччя життя.
Супутні порушення інших органів та систем:
З боку опорно-рухового апарату часто бувають сколіози, кіфози, деформації грудної клітки. Остеопороз зустрічається у більше ніж 90% випадків. Витончення кортикального шару довгих трубчастих кісток може призводити до формування несправжніх суглобів.
Найхарактернішими психічними порушеннями є розумова відсталість, затримка мовного розвитку, дислексія, когнітивні порушення, які спостерігаються у 20-30% дітей. Інтелектуальна недостатність звичайно пов’язана з недостатністю пам’яті, уваги, динаміки психічних процесів.
Основним завданням наукових досліджень є розробка методів патогенетичного лікування нейрофіброматозу I типу, які б дали змогу стримувати появу нових і ріст існуючих пухлин, а також запобігати розвитку ускладнень.
Нейрофіброматоз II типу (НФ2):
НФ2 (центральний нейрофіброматоз, двобічний слуховий нейрофіброматоз, код по OMIM – 101000) – це прогресуюче інвалідизуюче моногенне аутосомно-домінантне захворювання, яке проявляється двобічними вестибулярними шванномами, а також множинними пухлинами центральної та периферичної нервової системи, ранніми катарактами. Зустрічається з частотою 1:200000 населення.
Міжнародним комітетом експертів з нейрофіброматозу при Національному інституті здоров’я США в 1987 році рекомендовані діагностичні критерії НФ2:
- Двобічна невринома слухового нерву, підтверджена МРТ.
- Наявність НФ2 у прямих родичів.
- Однобічна пухлина (новоутворення) VIII черепного нерву або
- Наявність двох з наступних ознак: нейрофіброма, менінгіома, гліома, шваннома, ювенільна задня субкапсулярна катаракта.
Ген НФ2 знаходиться в 22 хромосомі (22q12). Продукт гену – білок мерлін (шванномін) є білком-супресором пухлин. Мерлін по структурі та властивостях дуже близький до трьох гомологічних білків: моезіну, езріну і радіксіну. Найбільше значення вони мають в регулюванні проліферації клітин нейроектодермального походження. Детально патогенез захворювання на сьогоднішній день не досліджений, однак багато авторів вказують на схожі ланки етіопатогенезу при НФ2 і спорадичних менінгіомах та шванномах.
Симптоми НФ2 звичайно з’являються під час другого десятиріччя життя або пізніше.
Двобічна невринома слухових нервів реєструється у більше ніж 90% хворих. Захворювання маніфестує, як правило, шумом у вухах, зниженням слуху. Крім того, зустрічаються менінгіоми, шванноми, епендімоми головного та спинного мозку, черепно-мозкових нервів. При цьому з’являються скарги на порушення координації, ністагм, головні болі. Можуть розвиватися судомні напади.
Плями кольору “кави з молоком” і периферичні нейрофіброми зустрічаються також у хворих на НФ2, але рідше, ніж при НФ1. Звичайно їх буває менше 6.
Нейрофіброматоз III типу:
Нейрофіброматоз III типу (код по OMIM – 162260) – долонні нейрофіброми, бліді відносно великі плями кольору кави з молоком, двобічні невроми слухового нерву, менінгіоми задньої ямки та верхнього шийного відділу, спінальні та параспінальні нейрофіброми; відсутність вузликів Ліша на райдужці; пухлини центральної нервової системи, що швидко розвиваються під час другого і третього десятиріччя життя.
Нейрофіброматоз IV типу:
Нейрофіброматоз IV типу (код по OMIM – 162270) – клініка нейрофіброматозу I типу, але без вузликів Ліша.
Лікування:
Радикального методу лікування нейрофіброматозу поки що не існує. Вчені сконцентрувались на можливості етіологічного лікування, тобто генної інженерії. Особливо далеко цей напрямок просунувся з моменту відкриття мутантного гену та розшифровки його первинного продукту — нейрофіброміну в 1990 році. На наукові дослідження, пов’язані з цією проблемою, щорічно виділяються величезні кошти, однак результатів, які можна було впровадити в практичну медицину, поки не досягнуто.
Для лікування цього захворювання традиційно використовуються методи симптоматичної терапії: хірургічне видалення пухлин або променева терапія нейрофібром внутрішніх органів. Проте ці методи не можна використовувати в якості методів вибору, враховуючи множинність пухлин, а також можливість рецидиву та прискорення росту інших нейрофібром після хірургічного втручання. Це ж можна сказати й про використання лазерної хірургічної методики видалення усіх нейрофібром на поверхні шкіри та в підшкірному шарі хворого, тому що це не рятує від виникнення нових чи посиленого росту існуючих гамартом в центральній нервовій системі. Вочевидь, хірургічні методи лікування нейрофіброматозу повинні застосовуватись за суворими показами: болі, порушення чутливості та рухових функцій, швидкий ріст пухлини, з обґрунтуванням термінів операції.
Перша спроба патогенетичного підходу до лікування була зроблена V. Riccardi в 1987 році, коли він запропонував довготривале використання кетотіфену (в дозі 2-4 мг протягом 1,5-3 років) для стабілізації мембран тучних клітин, вважаючи, що саме дегрануляція цих клітин стимулює ріст пухлин. Проте лікування одним кетотіфеном не принесло очікуваних результатів: зменшувались суб’єктивні відчуття болю і свербіння в ділянці нейрофібром, але якого-небудь впливу на ріст пухлин відмічено не було.
З того часу багато вітчизняних і закордонних авторів пропонували свої методи медикаментозної терапії, направленої на запобігання появі нових нейрофібром і сповільнення зростання існуючих. Так, в Центральному науково-дослідному шкірно-венерологічному інституті (Росія) у відділі спадкових захворювань шкіри розроблена методика лікування хворих на нейрофіброматоз, яка включає препарати, що гальмують дегрануляцію тучних клітин (кетотіфен та його аналоги), знижують рівень в тканинах глікозамінгліканів (лідаза та її аналоги), засоби з антипроліферативним ефектом (тігазон, вітамін А у великих дозах). Аналогічні схеми лікування пропонуються й іншими авторами з ширшим використанням системних ензимних препаратів, препаратів-інгібіторів синтезу ДНК, інгібіторів синтезу цитокінів та інш. Вищевказані препарати використовуються комплексно в різних комбінаціях або у вигляді монотерапії залежно від форми нейрофіброматозу, скарг, перебігу, а також віку і статі хворих. Обов’язково лікування проводилось під час прогресування захворювання, тобто при появі нових пухлин та/або рості існуючих, які, як правило, супроводжуються свербінням або відчуттям болю в їх проекції, а також з метою запобігання активізації хвороби під час запланованих операцій на пухлинах.
Нижче наведено рекомендації Американської академії педіатрії щодо медичного нагляду за дітьми з нейрофіброматозом (pdf-файл).
Медичний нагляд за дітьми з нейрофіброматозом
(Схвалено Американською педіатричною академією)
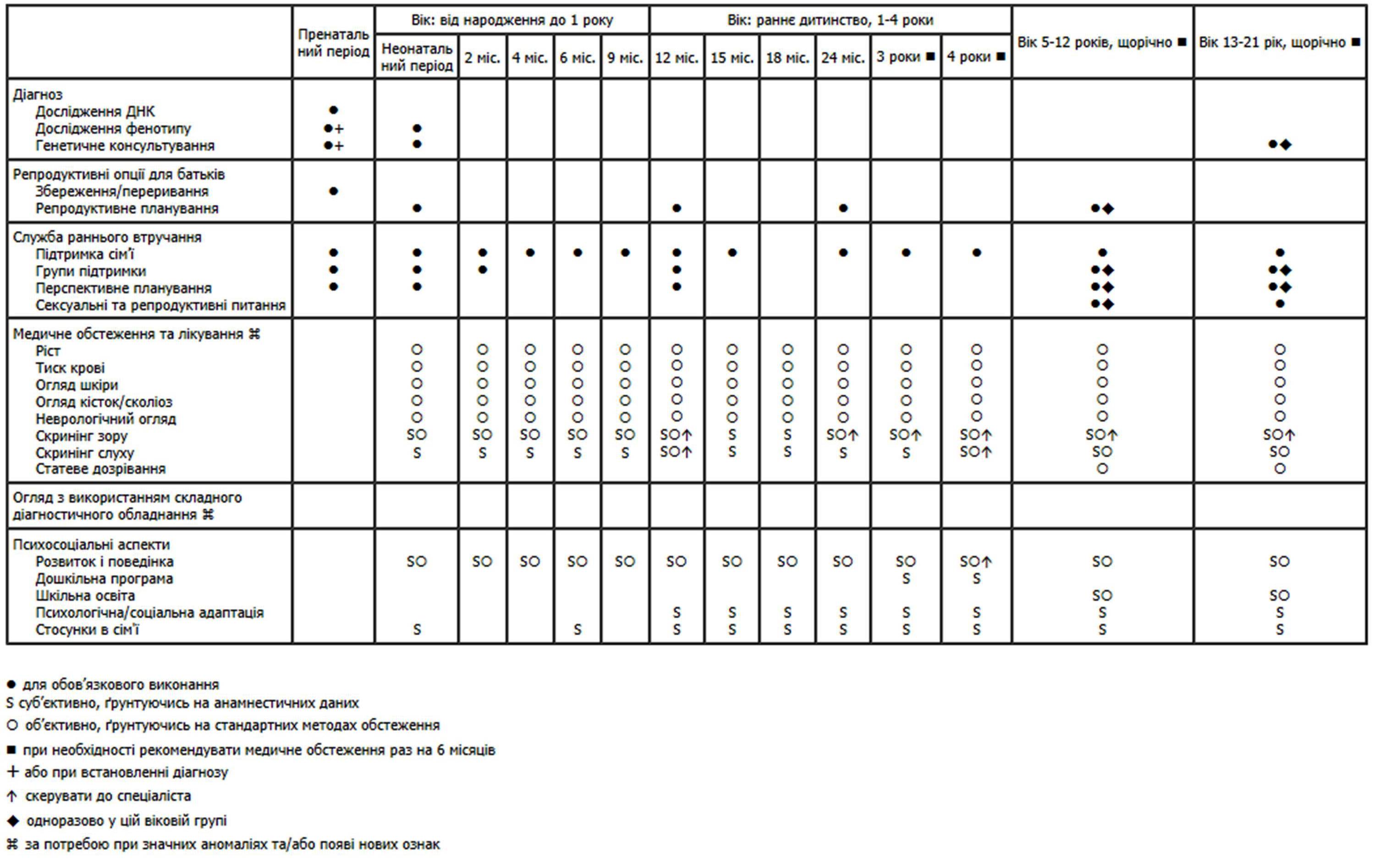
Література:
- Каталинич Д. /Хирургическое лечение нейрофиброматоза с помощью лазера. //Хирургия. – № 5. – 1996.
- Козлов А.В. /Наследственные заболевания в нейроонкологии. //Вопросы нейрохирургии. – № 3. – 2003.
- Короленко В.В., Бондур В.В. /Фармакотерапія нейрофіброматозу I типу як альтернативний метод лікування: сучасні досягнення і перспективи розвитку. //Дерматологія та венерологія. – № 4(18). – 2002.
- Макурдумян Л.А. /Нейрофиброматоз I типа. Проблемы диагностики и лечения. //Лечащий врач. – № 10. – 2001.
- Михайловский М.В. /Деформации позвоночника при нейрофиброматозе: обзор литературы. //Хирургия позвоночника. – № 3. – 2005.
- Мордовцева В.В., Мордовцева В.В. /Нейрофиброматоз I типа (болезнь Реклингаузена): ген открыт, клинико-патогенетические и терапевтические проблемы остаются. //Российский журнал кожных и венерических болезней. – № 2. – 1999.
- Цимбалюк В.І, Квасніцький М.В. /Діагностика та лікування нейрофіброматозу. //Мистецтво лікування. – № 5 (011). – 2004.
- Health Supervision for Children With Neurofibromatosis. American Academy of Pediatrics policy (http://aappolicy.aappublications.org).
- Packer RJ, Rosser T. Therapy for plexiform neurofibromas in children with neurofibromatosis 1: an overview. J Child Neurol. 2002 Aug;17(8):638-41.
Переглянуто редакційною колегією I.B.I.S.: 15/11/2005
Дивіться також:
- Нейрофіброматоз (інформація для батьків)
|
|
|
|
|
|




